map¶
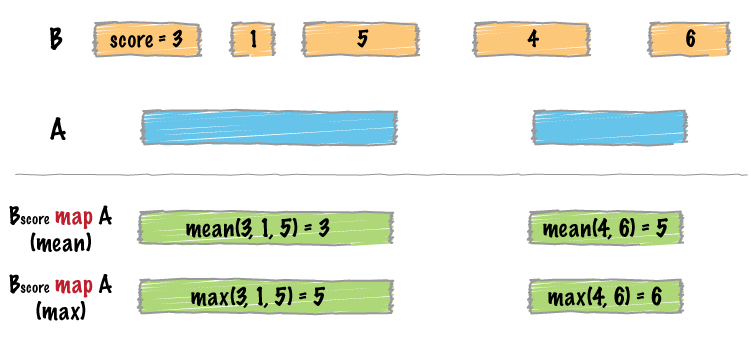
bedtools map allows one to map overlapping features in a B file onto features in an A file and apply statistics and/or summary operations on those features.
For example, one could use bedtools map to compute the average score of BEDGRAPH records that overlap genes. Since the fourth column in BEDGRAPH is the score, the following command illustrates how this would be done:
$ bedtools map -a genes.bed -b peaks.bedgraph -c 4 -o mean
Another example is discussed in this Biostars post.
Note
bedtools map requires each input file to be sorted by genome coordinate. For BED files, this can be done with sort -k1,1 -k2,2n. Other sorting criteria are allowed if a genome file (-g) is provides that specifies the expected chromosome order.
Note
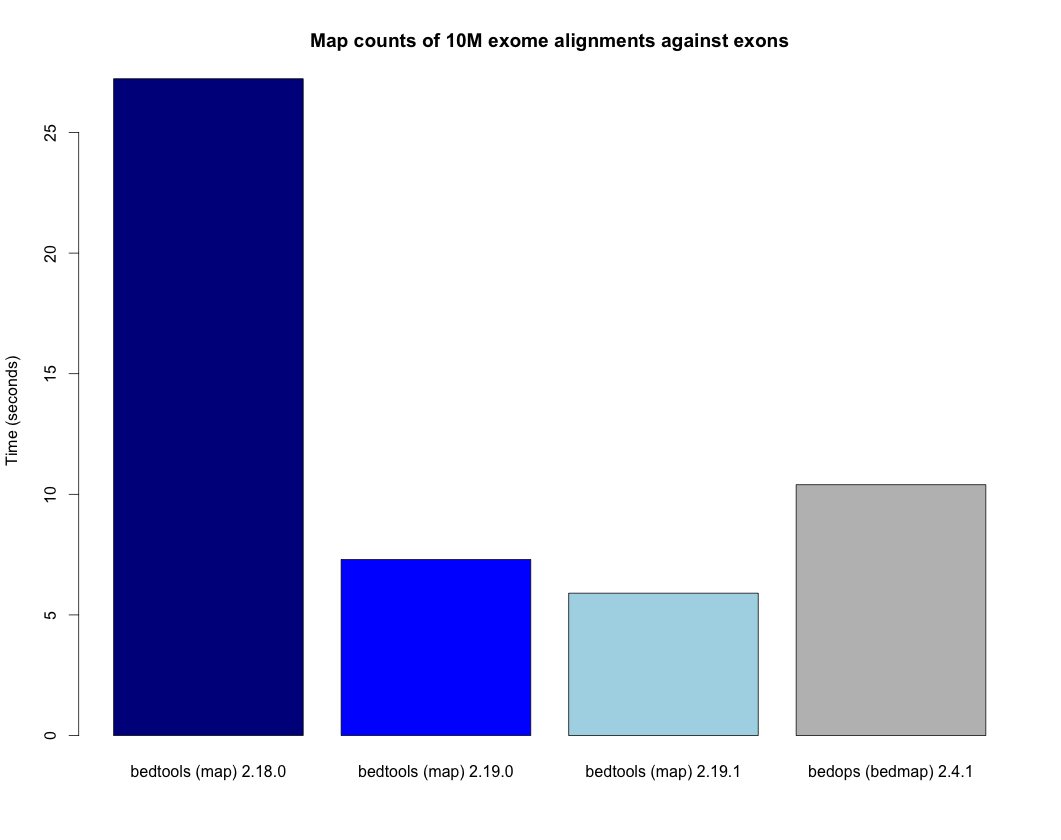
The map tool is substantially faster in versions 2.19.0 and later. The plot below demonstrates the increased speed when, for example, counting the number of exome alignments that align to each exon. The bedtools times are compared to the bedops bedmap utility as a point of reference.
Usage and option summary¶
Usage:
bedtools map [OPTIONS] -a <bed/gff/vcf> -b <bed/gff/vcf>
(or):
mapBed [OPTIONS] -a <bed/gff/vcf> -b <bed/gff/vcf>
| Option | Description |
|---|---|
| -c | Specify the column from the B file to map onto intervals in A.
Default: 5
|
| -o | Specify the operation that should be applied to -c. Valid operations:
sum - numeric only
count - numeric or text
count_distinct - numeric or text
min - numeric only
max - numeric only
absmin - numeric only
absmax - numeric only
mean - numeric only
median - numeric only
antimode - numeric or text
collapse (i.e., print a comma separated list) - numeric or text
distinct (i.e., print a comma separated list) - numeric or text
concat (i.e., print a comma separated list) - numeric or text
|
| -f | Minimum overlap required as a fraction of A. Default is 1E-9 (i.e. 1bp). |
| -F | Minimum overlap required as a fraction of B. Default is 1E-9 (i.e., 1bp). |
| -r | Require that the fraction of overlap be reciprocal for A and B. In other words, if -f is 0.90 and -r is used, this requires that B overlap at least 90% of A and that A also overlaps at least 90% of B. |
| -e | Require that the minimum fraction be satisfied for A _OR_ B. In other words, if -e is used with -f 0.90 and -F 0.10 this requires that either 90% of A is covered OR 10% of B is covered. Without -e, both fractions would have to be satisfied. |
| -s | Force “strandedness”. That is, only report hits in B that overlap A on the same strand. By default, overlaps are reported without respect to strand. |
| -S | Require different strandedness. That is, only report hits in B that overlap A on the _opposite_ strand. By default, overlaps are reported without respect to strand. |
| -null | The value to print if no overlaps are found for an A interval.
Default: "."
|
| -header | Print the header from the A file prior to results. |
| -split | Treat “split” BAM (i.e., having an “N” CIGAR operation) or BED12 entries as distinct BED intervals. When using -sorted, memory usage remains low even for very large files. |
| -g | Specify a genome file the defines the expected chromosome order in the input files. |
Default behavior - compute the sum of the score column for all overlaps.¶
By default, map computes the sum of the 5th column (the score field for BED format) for all intervals in B that overlap each interval in A.
Tip
Records in A that have no overlap will, by default, return . for the computed value from B. This can be changed with the -null option.
$ cat a.bed
chr1 10 20 a1 1 +
chr1 50 60 a2 2 -
chr1 80 90 a3 3 -
$ cat b.bed
chr1 12 14 b1 2 +
chr1 13 15 b2 5 -
chr1 16 18 b3 5 +
chr1 82 85 b4 2 -
chr1 85 87 b5 3 +
$ bedtools map -a a.bed -b b.bed
chr1 10 20 a1 1 + 12
chr1 50 60 a2 2 - .
chr1 80 90 a3 3 - 5
mean Compute the mean of a column from overlapping intervals¶
$ cat a.bed
chr1 10 20 a1 1 +
chr1 50 60 a2 2 -
chr1 80 90 a3 3 -
$ cat b.bed
chr1 12 14 b1 2 +
chr1 13 15 b2 5 -
chr1 16 18 b3 5 +
chr1 82 85 b4 2 -
chr1 85 87 b5 3 +
$ bedtools map -a a.bed -b b.bed -c 5 -o mean
chr1 10 20 a1 1 + 4
chr1 50 60 a2 2 - .
chr1 80 90 a3 3 - 2.5
collapse List each value of a column from overlapping intervals¶
$ bedtools map -a a.bed -b b.bed -c 5 -o collapse
chr1 10 20 a1 1 + 2,5,5
chr1 50 60 a2 2 - .
chr1 80 90 a3 3 - 2,3
distinct List each unique value of a column from overlapping intervals¶
$ bedtools map -a a.bed -b b.bed -c 5 -o distinct
chr1 10 20 a1 1 + 2,5
chr1 50 60 a2 2 - .
chr1 80 90 a3 3 - 2,3
-s Only include intervals that overlap on the same strand.¶
$ bedtools map -a a.bed -b b.bed -c 5 -o collapse -s
chr1 10 20 a1 1 + 2,5
chr1 50 60 a2 2 - .
chr1 80 90 a3 3 - 2
-S Only include intervals that overlap on the opposite strand.¶
$ bedtools map -a a.bed -b b.bed -c 5 -o collapse -S
chr1 10 20 a1 1 + 5
chr1 50 60 a2 2 - .
chr1 80 90 a3 3 - 3
Multiple operations and columns at the same time.¶
As of version 2.19.1, multiple columns and operations are allowed at the same time in a single run. This greatly expedites analyses by preventing one from having to process the same file over an over for each column/operation.
$ bedtools map -a a.bed -b b.bed -c 5,5,5,5 -o min,max,median,collapse
Or, apply the same function to multiple columns:
$ bedtools map -a a.bed -b b.bed -c 3,4,5,6 -o mean

Table Of Contents
- map
- Usage and option summary
- Default behavior - compute the sum of the score column for all overlaps.
- mean Compute the mean of a column from overlapping intervals
- collapse List each value of a column from overlapping intervals
- distinct List each unique value of a column from overlapping intervals
- -s Only include intervals that overlap on the same strand.
- -S Only include intervals that overlap on the opposite strand.
- Multiple operations and columns at the same time.