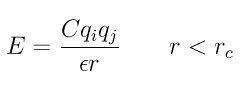
Syntax:
pair_style coul/cut cutoff pair_style coul/debye kappa cutoff pair_style coul/dsf alpha cutoff pair_style coul/long cutoff pair_style coul/long/gpu cutoff pair_style coul/wolf alpha cutoff pair_style tip4p/cut otype htype btype atype qdist cutoff pair_style tip4p/long otype htype btype atype qdist cutoff
Examples:
pair_style coul/cut 2.5 pair_coeff * * pair_coeff 2 2 3.5
pair_style coul/debye 1.4 3.0 pair_coeff * * pair_coeff 2 2 3.5
pair_style coul/dsf 0.05 10.0 pair_coeff * *
pair_style coul/long 10.0 pair_coeff * *
pair_style coul/msm 10.0 pair_coeff * *
pair_style coul/wolf 0.2 9.0 pair_coeff * *
pair_style tip4p/cut 1 2 7 8 0.15 12.0 pair_coeff * *
pair_style tip4p/long 1 2 7 8 0.15 10.0 pair_coeff * *
Description:
The coul/cut style computes the standard Coulombic interaction potential given by
where C is an energy-conversion constant, Qi and Qj are the charges on the 2 atoms, and epsilon is the dielectric constant which can be set by the dielectric command. The cutoff Rc truncates the interaction distance.
Style coul/debye adds an additional exp() damping factor to the Coulombic term, given by
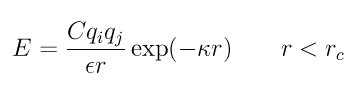
where kappa is the Debye length. This potential is another way to mimic the screening effect of a polar solvent.
Style coul/dsf computes Coulombic interactions via the damped shifted force model described in Fennell, given by:
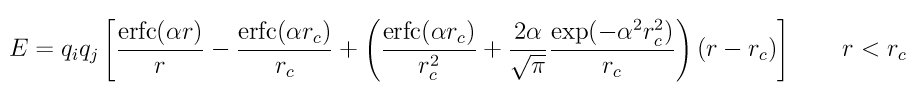
where alpha is the damping parameter and erfc() is the complementary error-function. The potential corrects issues in the Wolf model (described below) to provide consistent forces and energies (the Wolf potential is not differentiable at the cutoff) and smooth decay to zero.
Style coul/wolf computes Coulombic interactions via the Wolf summation method, described in Wolf, given by:
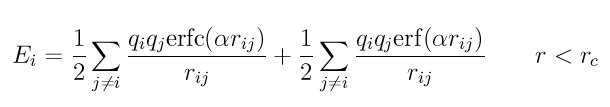
where alpha is the damping parameter, and erc() and erfc() are error-fuction and complementary error-function terms. This potential is essentially a short-range, spherically-truncated, charge-neutralized, shifted, pairwise 1/r summation. With a manipulation of adding and substracting a self term (for i = j) to the first and second term on the right-hand-side, respectively, and a small enough alpha damping parameter, the second term shrinks and the potential becomes a rapidly-converging real-space summation. With a long enough cutoff and small enough alpha parameter, the energy and forces calcluated by the Wolf summation method approach those of the Ewald sum. So it is a means of getting effective long-range interactions with a short-range potential.
Styles coul/long and coul/msm compute the same Coulombic interactions as style coul/cut except that an additional damping factor is applied so it can be used in conjunction with the kspace_style command and its ewald or pppm option. The Coulombic cutoff specified for this style means that pairwise interactions within this distance are computed directly; interactions outside that distance are computed in reciprocal space.
Styles tip4p/cut and tip4p/long implement the coulomb part of the TIP4P water model of (Jorgensen), which introduces a massless site located a short distance away from the oxygen atom along the bisector of the HOH angle. The atomic types of the oxygen and hydrogen atoms, the bond and angle types for OH and HOH interactions, and the distance to the massless charge site are specified as pair_style arguments. Style tip4p/cut uses a global cutoff for Coulomb interactions; style tip4p/long is for use with a long-range Coulombic solver (Ewald or PPPM).
IMPORTANT NOTE: For each TIP4P water molecule in your system, the atom IDs for the O and 2 H atoms must be consecutive, with the O atom first. This is to enable LAMMPS to "find" the 2 H atoms associated with each O atom. For example, if the atom ID of an O atom in a TIP4P water molecule is 500, then its 2 H atoms must have IDs 501 and 502.
See the howto section for more information on how to use the TIP4P pair styles and lists of parameters to set. Note that the neighobr list cutoff for Coulomb interactions is effectively extended by a distance 2*qdist when using the TIP4P pair style, to account for the offset distance of the fictitious charges on O atoms in water molecules. Thus it is typically best in an efficiency sense to use a LJ cutoff >= Coulomb cutoff + 2*qdist, to shrink the size of the neighbor list. This leads to slightly larger cost for the long-range calculation, so you can test the trade-off for your model.
These potentials are designed to be combined with other pair potentials via the pair_style hybrid/overlay command. This is because they have no repulsive core. Hence if they are used by themselves, there will be no repulsion to keep two oppositely charged particles from overlapping each other.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the examples above, or in the data file or restart files read by the read_data or read_restart commands, or by mixing as described below:
For coul/cut and coul/debye, the cutoff coefficient is optional. If it is not used (as in some of the examples above), the default global value specified in the pair_style command is used.
For coul/long and coul/msm no cutoff can be specified for an individual I,J type pair via the pair_coeff command. All type pairs use the same global Coulombic cutoff specified in the pair_style command.
Styles with a cuda, gpu, omp, or opt suffix are functionally the same as the corresponding style without the suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section_accelerate of the manual. The accelerated styles take the same arguments and should produce the same results, except for round-off and precision issues.
These accelerated styles are part of the USER-CUDA, GPU, USER-OMP and OPT packages, respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your input script.
See Section_accelerate of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the cutoff distance for the coul/cut style can be mixed. The default mix value is geometric. See the "pair_modify" command for details.
The pair_modify shift option is not relevant for these pair styles.
The coul/long style supports the pair_modify table option for tabulation of the short-range portion of the long-range Coulombic interaction.
These pair styles do not support the pair_modify tail option for adding long-range tail corrections to energy and pressure.
These pair styles write their information to binary restart files, so pair_style and pair_coeff commands do not need to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the inner, middle, outer keywords.
Restrictions:
The coul/long, coul/msm and tip4p/long styles are part of the KSPACE package. They are only enabled if LAMMPS was built with that package (which it is by default). See the Making LAMMPS section for more info.
Related commands:
pair_coeff, pair_style hybrid/overlay kspace_style
Default: none
(Wolf) D. Wolf, P. Keblinski, S. R. Phillpot, J. Eggebrecht, J Chem Phys, 110, 8254 (1999).
(Fennell) C. J. Fennell, J. D. Gezelter, J Chem Phys, 124, 234104 (2006).