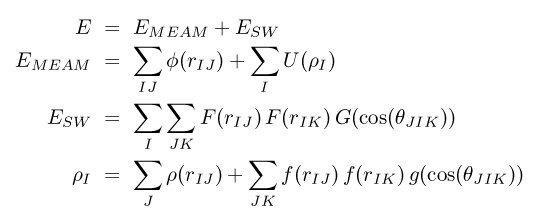
Syntax:
pair_style meam/sw/spline
Examples:
pair_style meam/sw/spline pair_coeff * * Ti.meam.sw.spline Ti pair_coeff * * Ti.meam.sw.spline Ti Ti Ti
Description:
The meam/sw/spline style computes pairwise interactions for metals using a variant of modified embedded-atom method (MEAM) potentials (Lenosky) with an additional Stillinger-Weber (SW) term (Stillinger) in the energy. This form of the potential was first proposed by Nicklas, Fellinger, and Park (Nicklas). We refer to it as MEAM+SW. The total energy E is given by
where rho_I is the density at atom I, theta_JIK is the angle between atoms J, I, and K centered on atom I. The seven functions Phi, F, G, U, rho, f, and g are represented by cubic splines.
The cutoffs and the coefficients for these spline functions are listed in a parameter file which is specified by the pair_coeff command. Parameter files for different elements are included in the "potentials" directory of the LAMMPS distribution and have a ".meam.sw.spline" file suffix. All of these files are parameterized in terms of LAMMPS metal units.
Note that unlike for other potentials, cutoffs for spline-based MEAM+SW potentials are not set in the pair_style or pair_coeff command; they are specified in the potential files themselves.
Unlike the EAM pair style, which retrieves the atomic mass from the potential file, the spline-based MEAM+SW potentials do not include mass information; thus you need to use the mass command to specify it.
Only a single pair_coeff command is used with the meam/sw/spline style which specifies a potential file with parameters for all needed elements. These are mapped to LAMMPS atom types by specifying N additional arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom types:
See the pair_coeff doc page for alternate ways to specify the path for the potential file.
As an example, imagine the Ti.meam.sw.spline file has values for Ti. If your LAMMPS simulation has 3 atoms types and they are all to be treated with this potential, you would use the following pair_coeff command:
pair_coeff * * Ti.meam.sw.spline Ti Ti Ti
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The three Ti arguments map LAMMPS atom types 1,2,3 to the Ti element in the potential file. If a mapping value is specified as NULL, the mapping is not performed. This can be used when a meam/sw/spline potential is used as part of the hybrid pair style. The NULL values are placeholders for atom types that will be used with other potentials.
IMPORTANT NOTE: The meam/sw/spline style currently supports only single-element MEAM+SW potentials. It may be extended for alloy systems in the future.
Example input scripts that use this pair style are provided in the examples/USER/misc/meam_sw_spline directory.
Mixing, shift, table, tail correction, restart, rRESPA info:
The pair style does not support multiple element types or mixing. It has been designed for pure elements only.
This pair style does not support the pair_modify shift, table, and tail options.
The meam/sw/spline pair style does not write its information to binary restart files, since it is stored in an external potential parameter file. Thus, you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
The meam/sw/spline pair style can only be used via the pair keyword of the run_style respa command. They do not support the inner, middle, outer keywords.
Restrictions:
This pair style requires the newton setting to be "on" for pair interactions.
This pair style is only enabled if LAMMPS was built with the USER-MISC package. See the Making LAMMPS section for more info.
Related commands:
pair_coeff, pair_style meam, pair_style meam/spline
Default: none
(Lenosky) Lenosky, Sadigh, Alonso, Bulatov, de la Rubia, Kim, Voter, Kress, Modell. Simul. Mater. Sci. Eng. 8, 825 (2000).
(Stillinger) Stillinger, Weber, Phys. Rev. B 31, 5262 (1985).
(Nicklas) The spline-based MEAM+SW format was first devised and used to develop potentials for bcc transition metals by Jeremy Nicklas, Michael Fellinger, and Hyoungki Park at The Ohio State University.