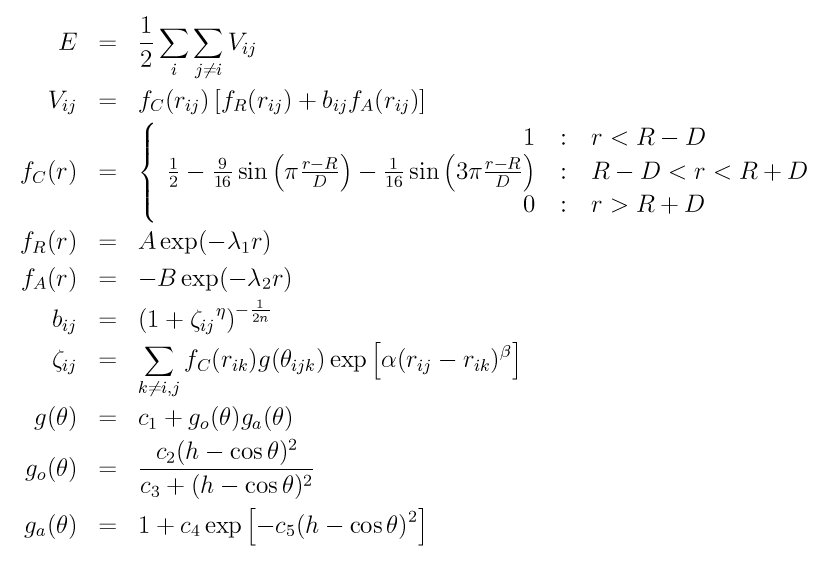
Syntax:
pair_style tersoff/mod
Examples:
pair_style tersoff/mod pair_coeff * * Si.tersoff.mod Si Si
Description:
The tersoff/mod style computes a bond-order type interatomic potential (Kumagai) based on a 3-body Tersoff potential (Tersoff_1), (Tersoff_2) with modified cutoff function and angular-dependent term, giving the energy E of a system of atoms as
where f_R is a two-body term and f_A includes three-body interactions. The summations in the formula are over all neighbors J and K of atom I within a cutoff distance = R + D.
The modified cutoff function f_C proposed by (Murty) and having a continuous second-order differential is employed. The angular-dependent term g(theta) was modified to increase the flexibility of the potential.
The tersoff/mod potential is fitted to both the elastic constants and melting point by employing the modified Tersoff potential function form in which the angular-dependent term is improved. The model performs extremely well in describing the crystalline, liquid, and amorphous phases (Schelling).
Only a single pair_coeff command is used with the tersoff/mod style which specifies a Tersoff/MOD potential file with parameters for all needed elements. These are mapped to LAMMPS atom types by specifying N additional arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom types:
As an example, imagine the Si.tersoff_mod file has Tersoff values for Si. If your LAMMPS simulation has 3 Si atoms types, you would use the following pair_coeff command:
pair_coeff * * Si.tersoff_mod Si Si Si
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The three Si arguments map LAMMPS atom types 1,2,3 to the Si element in the Tersoff/MOD file. If a mapping value is specified as NULL, the mapping is not performed. This can be used when a tersoff/mod potential is used as part of the hybrid pair style. The NULL values are placeholders for atom types that will be used with other potentials.
Tersoff/MOD file in the potentials directory of the LAMMPS distribution have a ".tersoff.mod" suffix. Lines that are not blank or comments (starting with #) define parameters for a triplet of elements. The parameters in a single entry correspond to coefficients in the formula above:
The n, eta, lambda2, B, lambda1, and A parameters are only used for two-body interactions. The beta, alpha, c1, c2, c3, c4, c5, h parameters are only used for three-body interactions. The R and D parameters are used for both two-body and three-body interactions. The non-annotated parameters are unitless.
The Tersoff/MOD potential file must contain entries for all the elements listed in the pair_coeff command. It can also contain entries for additional elements not being used in a particular simulation; LAMMPS ignores those entries.
For a single-element simulation, only a single entry is required (e.g. SiSiSi). As annotated above, the first element in the entry is the center atom in a three-body interaction and it is bonded to the 2nd atom and the bond is influenced by the 3rd atom. Thus an entry for SiSiSi means Si bonded to a Si with another Si atom influencing the bond.
Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support the pair_modify shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in potential files. Thus, you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the inner, middle, outer keywords.
Restrictions:
This pair style is part of the MANYBODY package. It is only enabled if LAMMPS was built with that package (which it is by default). See the Making LAMMPS section for more info.
This pair style requires the newton setting to be "on" for pair interactions.
The Tersoff/MOD potential files provided with LAMMPS (see the potentials directory) are parameterized for metal units. You can use the Tersoff/MOD potential with any LAMMPS units, but you would need to create your own Tersoff/MOD potential file with coefficients listed in the appropriate units if your simulation doesn't use "metal" units.
Related commands:
Default: none
(Kumagai) T. Kumagai, S. Izumi, S. Hara, S. Sakai, Comp. Mat. Science, 39, 457 (2007).
(Tersoff_1) J. Tersoff, Phys Rev B, 37, 6991 (1988).
(Tersoff_2) J. Tersoff, Phys Rev B, 38, 9902 (1988).
(Murty) M.V.R. Murty, H.A. Atwater, Phys Rev B, 51, 4889 (1995).
(Schelling) Patrick K. Schelling, Comp. Mat. Science, 44, 274 (2008).