


Next: Sample foldings
Up: Running the programs
Previous: Optimal and suboptimal foldings
The mfold package contains a script, also named mfold , that performs
a folding according to information entered on the command line. This
script is itself composed of scripts and (Fortran or C) executable
programs, many of which can be run separately. Some of these programs
are now described.
- 1.
- auxgen: This program creates the `FILE_NAME.PNT' file.
- 2.
- boxplot97_ng: This program creates an energy dot plot in PostScript or
gif form from `FILE_NAME.PLOT'. The file BOXPLOT97_NG.DOC
contains instructions.
- 3.
- ct_boxplot: This program creates a dot plot containing
only those base pairs found in a collection of structures (in ``ct
format'') that are specified on the command line. These must all be
foldings of the same sequence fragment. At present, the mfold script
does not use this program.
- 4.
- ct_compare: This program compares 2 ``ct files''. The
first contains a single reference structure. The second contains 1 or
more foldings of the same sequence. The number of bases and helices
from the first structure that are conserved in the other structures
are computed and displayed. For the purposes of this program, a
``helix'' may contain bulge or interior loops of size 1 or 2 and must
have at least 3 base pairs [17,40].
- 5.
- efn: This program computes
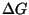 for all the foldings in a
``ct file''. The energy rules correspond exactly to what is used in mfold .
for all the foldings in a
``ct file''. The energy rules correspond exactly to what is used in mfold .
- 6.
- efn2: This program computes
for all the foldings in
a ``ct file'' using a more precise free energy computation that takes
into account coaxial stacking and Jacobson-Stockmeyer theory for
multi-branched loops. (See equation 7 and Figure 8,
respectively.)
- 7.
- h-num: This program computes an ``h-num'' file from a
``plot'' file.
- 8.
- nafold: This is the principle folding program of the
mfold package, and corresponds to the program ``lrna'' and ``crna''
in earlier versions of mfold . It has 2 command line arguments. The
first takes on the values `l' or `c', for linear (default) or
circular folding, respectively. The second has values ``text''
(default) or ``html'' for plain text output, and for some html
output, as described above. It is run twice by the mfold script. It
may be run alone, as it prompts the user for input on an interactive
basis. In this mode, the user may fold all the sequences contained in
a single file, an option not available with the mfold script. The
instructions for running this program have been described in
[19] and also on the WWW at
http://www.bioinfo.rpi.edu/
~zukerm/mfold-2.0.
The interactive energy dot plot is not functional in version 3.0.
- 9.
- naview: This is a modified version of the naview
program described in [41]. Actually, in mfold , naview is a script that runs a binary called naview.exe. Both
naview and naview.exe may be run alone in interactive
form. The mfold script uses the files ``bases.nav'' and
``lines.nav'', stored in MFOLDLIB to direct ``naview''. The
first produces an output that displays individual residues as letters,
while the second gives a structure outline only.
- 10.
- newtemp: The newtemp program creates free energy
files for folding RNA or DNA at different temperatures. The latest RNA
parameters consist only of free energies measured at 37°.
Since there are no corresponding enthalpy files, it is not possible to
fold RNA at temperatures other than 37°.
If the user wishes
to fold RNA at different temperatures, then the ``dg'' and ``dh''
files from mfold version 2.3 should be copied into MFOLDLIB. These older free energy parameters will be supplied with
mfold version 3.0. DNA folding may be done at arbitrary temperatures
between 0 and 100 degrees. However, it should be remembered that the
DNA loop parameters are all estimated from values published in the
literature or by comparison with RNA.
- 11.
- plt22ps: This program takes a ``plt2'' file from naview and creates a PostScript file of a plotted structure. It can
use an ``ann'' file or an ``ss-count'' file to annotate with P-num or
ss-count values, respectively.
- 12.
- plt22gif: This program is the same as plt22ps,
except that the structure output file is in gif format.
- 13.
- sav2p-num: This interactive program uses an existing
`fold_name.sav' file to create a P-num file.
- 14.
- sav2plot: This interactive program uses an existing
`fold_name.sav' file to create a ``plot'' file. The resulting
``plot'' file may be sorted and filtered using the command:
FILTER-SORT NAME.PLOT N, where name.plot is a ``plot'' file, and
N is the minimum helix size that is desired. Helices shorter than
N and not removed in optimal foldings.
- 15.
- scorer: This program is similar to ct_compare. It
compares foldings helix by helix, displaying a more detailed output.
- 16.
- split_ct.awk: This is a Unix awk script that splits a ct
file into multiple files, containing 1 folding in each. These
individual files may be processed by naview, plt22ps and
plt22gif.
- 17.
- ss-count: This program computes single stranded sample
statistics from a collection of foldings stored in a single ct file.
Next: Sample foldings
Up: Running the programs
Previous: Optimal and suboptimal foldings
 | Michael Zuker
Institute for Biomedical Computing
Washington University in St. Louis
1998-12-05 |