H/part_func.h File Reference
Partition function of single RNA sequences. More...
Go to the source code of this file.
Functions | |
| float | pf_fold_par (const char *sequence, char *structure, pf_paramT *parameters, int calculate_bppm, int is_constrained, int is_circular) |
Compute the partition function 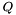 for a given RNA sequence. for a given RNA sequence. | |
| float | pf_fold (const char *sequence, char *structure) |
| Compute the partition function of an RNA sequence. | |
| float | pf_circ_fold (const char *sequence, char *structure) |
| Compute the partition function of a circular RNA sequence. | |
| char * | pbacktrack (char *sequence) |
| Sample a secondary structure from the Boltzmann ensemble according its probability . | |
| char * | pbacktrack_circ (char *sequence) |
| Sample a secondary structure of a circular RNA from the Boltzmann ensemble according its probability. | |
| void | free_pf_arrays (void) |
| Free arrays for the partition function recursions. | |
| void | update_pf_params (int length) |
| Recalculate energy parameters. | |
| FLT_OR_DBL * | export_bppm (void) |
| Get a pointer to the base pair probability array. | |
| void | assign_plist_from_pr (plist **pl, FLT_OR_DBL *probs, int length, double cutoff) |
| Create a plist from a probability matrix. | |
| int | get_pf_arrays (short **S_p, short **S1_p, char **ptype_p, FLT_OR_DBL **qb_p, FLT_OR_DBL **qm_p, FLT_OR_DBL **q1k_p, FLT_OR_DBL **qln_p) |
| Get the pointers to (almost) all relavant computation arrays used in partition function computation. | |
| double | get_subseq_F (int i, int j) |
| Get the free energy of a subsequence from the q[] array. | |
| char * | get_centroid_struct_pl (int length, double *dist, plist *pl) |
| Get the centroid structure of the ensemble. | |
| char * | get_centroid_struct_pr (int length, double *dist, FLT_OR_DBL *pr) |
| Get the centroid structure of the ensemble. | |
| double | mean_bp_distance (int length) |
| Get the mean base pair distance of the last partition function computation. | |
| double | mean_bp_distance_pr (int length, FLT_OR_DBL *pr) |
| Get the mean base pair distance in the thermodynamic ensemble. | |
| void | bppm_to_structure (char *structure, FLT_OR_DBL *pr, unsigned int length) |
| Create a dot-bracket like structure string from base pair probability matrix. | |
| char | bppm_symbol (const float *x) |
| Get a pseudo dot bracket notation for a given probability information. | |
| void | init_pf_fold (int length) |
| Allocate space for pf_fold(). | |
| char * | centroid (int length, double *dist) |
| double | mean_bp_dist (int length) |
| get the mean pair distance of ensemble | |
| double | expLoopEnergy (int u1, int u2, int type, int type2, short si1, short sj1, short sp1, short sq1) |
| double | expHairpinEnergy (int u, int type, short si1, short sj1, const char *string) |
Variables | |
| int | st_back |
| a flag indicating that auxilary arrays are needed throughout the computations which are necessary for stochastic backtracking | |
Detailed Description
Partition function of single RNA sequences.
This file includes (almost) all function declarations within the RNAlib that are related to Partion function folding...
- Note:
- If you plan on using the functions provided from this section of the RNAlib concurrently via OpenMP you have to place a COPYIN clause right before your PARALLEL directive! Otherwise, some functions may not behave as expected. A complete list of variables that have to be passed to the COPYIN clause can be found in the detailed description of each function below.
Function Documentation
| float pf_fold_par | ( | const char * | sequence, | |
| char * | structure, | |||
| pf_paramT * | parameters, | |||
| int | calculate_bppm, | |||
| int | is_constrained, | |||
| int | is_circular | |||
| ) |
Compute the partition function for a given RNA sequence.
If structure is not a NULL pointer on input, it contains on return a string consisting of the letters " . , | { } ( ) " denoting bases that are essentially unpaired, weakly paired, strongly paired without preference, weakly upstream (downstream) paired, or strongly up- (down-)stream paired bases, respectively. If fold_constrained is not 0, the structure string is interpreted on input as a list of constraints for the folding. The character "x" marks bases that must be unpaired, matching brackets " ( ) " denote base pairs, all other characters are ignored. Any pairs conflicting with the constraint will be forbidden. This is usually sufficient to ensure the constraints are honored. If tha parameter calculate_bppm is set to 0 base pairing probabilities will not be computed (saving CPU time), otherwise after calculations took place pr will contain the probability that bases i and j pair.
- Note:
- The global array pr is deprecated and the user who wants the calculated base pair probabilities for further computations is advised to use the function export_bppm()
- Parameters:
-
sequence The RNA sequence input structure A pointer to a char array where a base pair probability information can be stored in a pseudo-dot-bracket notation (may be NULL, too) parameters Data structure containing the precalculated Boltzmann factors calculate_bppm Switch to Base pair probability calculations on/off (0==off) is_constrained Switch to indicate that a structure contraint is passed via the structure argument (0==off) is_circular Switch to (de-)activate postprocessing steps in case RNA sequence is circular (0==off)
- Returns:
- The Gibbs free energy of the ensemble (
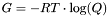 ) in kcal/mol
) in kcal/mol
| float pf_fold | ( | const char * | sequence, | |
| char * | structure | |||
| ) |
Compute the partition function of an RNA sequence.
If structure is not a NULL pointer on input, it contains on return a string consisting of the letters " . , | { } ( ) " denoting bases that are essentially unpaired, weakly paired, strongly paired without preference, weakly upstream (downstream) paired, or strongly up- (down-)stream paired bases, respectively. If fold_constrained is not 0, the structure string is interpreted on input as a list of constraints for the folding. The character "x" marks bases that must be unpaired, matching brackets " ( ) " denote base pairs, all other characters are ignored. Any pairs conflicting with the constraint will be forbidden. This is usually sufficient to ensure the constraints are honored. If do_backtrack has been set to 0 base pairing probabilities will not be computed (saving CPU time), otherwise pr will contain the probability that bases i and j pair.
- Note:
- The global array pr is deprecated and the user who wants the calculated base pair probabilities for further computations is advised to use the function export_bppm()
- See also:
- pf_circ_fold(), bppm_to_structure(), export_bppm()
- Parameters:
-
sequence The RNA sequence input structure A pointer to a char array where a base pair probability information can be stored in a pseudo-dot-bracket notation (may be NULL, too)
- Returns:
- The Gibbs free energy of the ensemble () in kcal/mol
| float pf_circ_fold | ( | const char * | sequence, | |
| char * | structure | |||
| ) |
Compute the partition function of a circular RNA sequence.
- See also:
- pf_fold(), pf_fold_par()
- Parameters:
-
sequence The RNA sequence input structure A pointer to a char array where a base pair probability information can be stored in a pseudo-dot-bracket notation (may be NULL, too)
- Returns:
- The Gibbs free energy of the ensemble () in kcal/mol
| char* pbacktrack | ( | char * | sequence | ) |
Sample a secondary structure from the Boltzmann ensemble according its probability
.
- Note:
- You have to call pf_fold() first in order to fill the partition function matrices
-
OpenMP notice:
This function relies on passing the following variables to the appropriate COPYIN clause (additionally to the ones needed by pf_fold()):
pstruc, sequence
- Parameters:
-
sequence The RNA sequence
- Returns:
- A sampled secondary structure in dot-bracket notation
| char* pbacktrack_circ | ( | char * | sequence | ) |
Sample a secondary structure of a circular RNA from the Boltzmann ensemble according its probability.
This function does the same as pbacktrack() but assumes the RNA molecule to be circular
- Note:
- OpenMP notice:
This function relies on passing the following variables to the appropriate COPYIN clause (additionally to the ones needed by pf_fold()):
pstruc, sequence
- Parameters:
-
sequence The RNA sequence
- Returns:
- A sampled secondary structure in dot-bracket notation
| void free_pf_arrays | ( | void | ) |
Free arrays for the partition function recursions.
Call this function if you want to free all allocated memory associated with the partition function forward recursion.
- Note:
- Successive calls of pf_fold(), pf_circ_fold() already check if they should free any memory from a previous run.
-
OpenMP notice:
This function should be called before leaving a thread in order to avoid leaking memory
- See also:
- pf_fold(), pf_circ_fold()
| void update_pf_params | ( | int | length | ) |
Recalculate energy parameters.
Call this function to recalculate the pair matrix and energy parameters after a change in folding parameters like temperature
| FLT_OR_DBL* export_bppm | ( | void | ) |
Get a pointer to the base pair probability array.
Accessing the base pair probabilities for a pair (i,j) is achieved by
FLT_OR_DBL *pr = export_bppm(); pr_ij = pr[iindx[i]-j];
- Note:
- Call pf_fold() before using this function!
- See also:
- pf_fold(), pf_circ_fold(), get_iindx()
- Returns:
- A pointer to the base pair probability array
| void assign_plist_from_pr | ( | plist ** | pl, | |
| FLT_OR_DBL * | probs, | |||
| int | length, | |||
| double | cutoff | |||
| ) |
Create a plist from a probability matrix.
The probability matrix given is parsed and all pair probabilities above the given threshold are used to create an entry in the plist
The end of the plist is marked by sequence positions i as well as j equal to 0. This condition should be used to stop looping over its entries
- Note:
- This function is threadsafe
| int get_pf_arrays | ( | short ** | S_p, | |
| short ** | S1_p, | |||
| char ** | ptype_p, | |||
| FLT_OR_DBL ** | qb_p, | |||
| FLT_OR_DBL ** | qm_p, | |||
| FLT_OR_DBL ** | q1k_p, | |||
| FLT_OR_DBL ** | qln_p | |||
| ) |
Get the pointers to (almost) all relavant computation arrays used in partition function computation.
- Note:
- In order to assign meaningful pointers, you have to call pf_fold first!
- See also:
- pf_fold(), pf_circ_fold()
- Parameters:
-
S_p A pointer to the 'S' array (integer representation of nucleotides) S1_p A pointer to the 'S1' array (2nd integer representation of nucleotides) ptype_p A pointer to the pair type matrix qb_p A pointer to the QB matrix qm_p A pointer to the QM matrix q1k_p A pointer to the 5' slice of the Q matrix ( 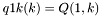 )
) qln_p A pointer to the 3' slice of the Q matrix ( 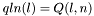 )
)
- Returns:
- Non Zero if everything went fine, 0 otherwise
| char* get_centroid_struct_pl | ( | int | length, | |
| double * | dist, | |||
| plist * | pl | |||
| ) |
Get the centroid structure of the ensemble.
This function is a threadsafe replacement for centroid() with a 'plist' input
The centroid is the structure with the minimal average distance to all other structures
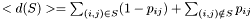
Thus, the centroid is simply the structure containing all pairs with 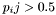 The distance of the centroid to the ensemble is written to the memory adressed by dist.
The distance of the centroid to the ensemble is written to the memory adressed by dist.
- Parameters:
-
length The length of the sequence dist A pointer to the distance variable where the centroid distance will be written to pl A pair list containing base pair probability information about the ensemble
- Returns:
- The centroid structure of the ensemble in dot-bracket notation
| char* get_centroid_struct_pr | ( | int | length, | |
| double * | dist, | |||
| FLT_OR_DBL * | pr | |||
| ) |
Get the centroid structure of the ensemble.
This function is a threadsafe replacement for centroid() with a probability array input
The centroid is the structure with the minimal average distance to all other structures
Thus, the centroid is simply the structure containing all pairs with The distance of the centroid to the ensemble is written to the memory adressed by dist.
- Parameters:
-
length The length of the sequence dist A pointer to the distance variable where the centroid distance will be written to pr A upper triangular matrix containing base pair probabilities (access via iindx get_iindx() )
- Returns:
- The centroid structure of the ensemble in dot-bracket notation
| double mean_bp_distance | ( | int | length | ) |
Get the mean base pair distance of the last partition function computation.
- Note:
- To ensure thread-safety, use the function mean_bp_distance_pr() instead!
- See also:
- mean_bp_distance_pr()
- Parameters:
-
length
- Returns:
- mean base pair distance in thermodynamic ensemble
| double mean_bp_distance_pr | ( | int | length, | |
| FLT_OR_DBL * | pr | |||
| ) |
Get the mean base pair distance in the thermodynamic ensemble.
This is a threadsafe implementation of mean_bp_dist() !
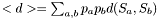
this can be computed from the pair probs 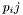 as
as
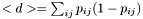
- Note:
- This function is threadsafe
- Parameters:
-
length The length of the sequence pr The matrix containing the base pair probabilities
- Returns:
- The mean pair distance of the structure ensemble
| void init_pf_fold | ( | int | length | ) |
Allocate space for pf_fold().
- Deprecated:
- This function is obsolete and will be removed soon!
| char* centroid | ( | int | length, | |
| double * | dist | |||
| ) |
- Deprecated:
- This function is deprecated and should not be used anymore as it is not threadsafe!
| double mean_bp_dist | ( | int | length | ) |
get the mean pair distance of ensemble
- Deprecated:
- This function is not threadsafe and should not be used anymore. Use mean_bp_distance() instead!
| double expLoopEnergy | ( | int | u1, | |
| int | u2, | |||
| int | type, | |||
| int | type2, | |||
| short | si1, | |||
| short | sj1, | |||
| short | sp1, | |||
| short | sq1 | |||
| ) |
- Deprecated:
- Use exp_E_IntLoop() from loop_energies.h instead
| double expHairpinEnergy | ( | int | u, | |
| int | type, | |||
| short | si1, | |||
| short | sj1, | |||
| const char * | string | |||
| ) |
- Deprecated:
- Use exp_E_Hairpin() from loop_energies.h instead
