5.1.38. window¶
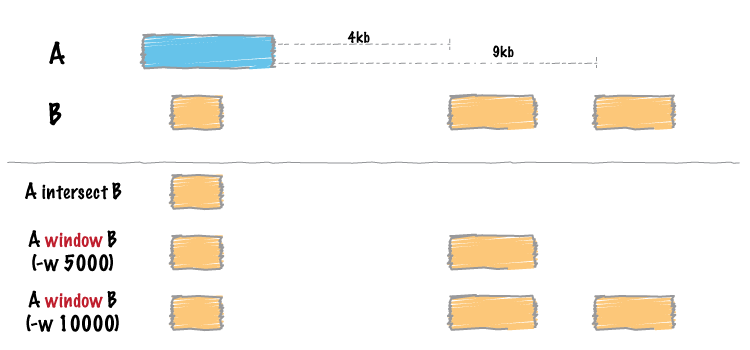
Similar to bedtools intersect, window searches for overlapping features in A and B. However, window adds a specified number (1000, by default) of base pairs upstream and downstream of each feature in A. In effect, this allows features in B that are “near” features in A to be detected.
5.1.38.1. Usage and option summary¶
Usage:
bedtools window [OPTIONS] [-a|-abam] -b <BED/GFF/VCF>
(or):
bedtools window [OPTIONS] [-a|-abam] -b <BED/GFF/VCF>
| Option | Description |
|---|---|
| -abam | BAM file A. Each BAM alignment in A is compared to B in search of overlaps. Use “stdin” if passing A with a UNIX pipe: For example: samtools view -b <BAM> | bedtools window -abam stdin -b genes.bed |
| -ubam | Write uncompressed BAM output. The default is write compressed BAM output. |
| -bed | When using BAM input (-abam), write output as BED. The default is to write output in BAM when using -abam. For example: bedtools window -abam reads.bam -b genes.bed -bed |
| -w | Base pairs added upstream and downstream of each entry in A when searching for overlaps in B. Default is 1000 bp. |
| -l | Base pairs added upstream (left of) of each entry in A when searching for overlaps in B. Allows one to create assymetrical “windows”. Default is 1000bp. |
| -r | Base pairs added downstream (right of) of each entry in A when searching for overlaps in B. Allows one to create assymetrical “windows”. Default is 1000bp. |
| -sw | Define -l and -r based on strand. For example if used, -l 500 for a negative-stranded feature will add 500 bp downstream. By default, this is disabled. |
| -sm | Only report hits in B that overlap A on the same strand. By default, overlaps are reported without respect to strand. |
| -Sm | Only report hits in B that overlap A on the opposite strand. By default, overlaps are reported without respect to strand. |
| -u | Write original A entry once if any overlaps found in B. In other words, just report the fact at least one overlap was found in B. |
| -c | For each entry in A, report the number of hits in B while restricting to -f. Reports 0 for A entries that have no overlap with B. |
| -v | Only report those entries in A that have no overlaps with B. |
| -header | Print the header from the A file prior to results. |
5.1.38.2. Default behavior¶
By default, bedtools window adds 1000 bp upstream and downstream of each A feature and searches for features in B that overlap this “window”. If an overlap is found in B, both the original A feature and the original B feature are reported.
$ cat A.bed
chr1 100 200
$ cat B.bed
chr1 500 1000
chr1 1300 2000
$ bedtools window -a A.bed -b B.bed
chr1 100 200 chr1 500 1000
5.1.38.3. -w Defining a custom window size¶
Instead of using the default window size of 1000bp, one can define a custom, symmetric window around each feature in A using the -w option. One should specify the window size in base pairs. For example, a window of 5kb should be defined as -w 5000.
For example (note that in contrast to the default behavior, the second B entry is reported):
$ cat A.bed
chr1 100 200
$ cat B.bed
chr1 500 1000
chr1 1300 2000
$ bedtools window -a A.bed -b B.bed -w 5000
chr1 100 200 chr1 500 1000
chr1 100 200 chr1 1300 2000
5.1.38.4. -l and -r Defining assymteric windows¶
One can also define asymmetric windows where a differing number of bases are added upstream and downstream of each feature using the -l (upstream) and -r (downstream)** options.
Note
By default, the -l and -r options ignore strand. If you want to define upstream and downstream based on strand, use the -sw option (below) with the -l and -r options.
For example (note the difference between -l 200 and -l 300):
$ cat A.bed
chr1 1000 2000
$ cat B.bed
chr1 500 800
chr1 10000 20000
$ bedtools window -a A.bed -b B.bed -l 200 -r 20000
chr1 1000 2000 chr1 10000 20000
$ bedtools window -a A.bed -b B.bed -l 300 -r 20000
chr1 1000 2000 chr1 500 800
chr1 1000 2000 chr1 10000 20000
5.1.38.5. -sw Defining assymteric windows based on strand¶
Especially when dealing with gene annotations or RNA-seq experiments, you may want to define asymmetric windows based on “strand”. For example, you may want to screen for overlaps that occur within 5000 bp upstream of a gene (e.g. a promoter region) while screening only 1000 bp downstream of the gene. By enabling the -sw (“stranded” windows) option, the windows are added upstream or downstream according to strand. For example, imagine one specifies -l 5000, -r 1000 as well as the -sw option. In this case, forward stranded (“+”) features will screen 5000 bp to the left (that is, lower genomic coordinates) and 1000 bp to the right (that is, higher genomic coordinates). By contrast, reverse stranded (“-”) features will screen 5000 bp to the right (that is, higher genomic coordinates) and 1000 bp to the left (that is, lower genomic coordinates).
For example (note the difference between -l 200 and -l 300):
$ cat A.bed
chr1 10000 20000 A.forward 1 +
chr1 10000 20000 A.reverse 1 -
$ cat B.bed
chr1 1000 8000 B1
chr1 24000 32000 B2
$ bedtools window -a A.bed -b B.bed -l 5000 -r 1000 -sw
chr1 10000 20000 A.forward 1 + chr1 1000 8000 B1
chr1 10000 20000 A.reverse 1 - chr1 24000 32000 B2
5.1.38.6. -sm Enforcing matches with the same “strandedness”¶
This option behaves the same as the -s option for bedtools intersect while scanning for overlaps within the “window” surrounding A. That is, overlaps in B will only be included if the B interval is on the same strand as the A interval.
5.1.38.7. -Sm Enforcing matches with the opposite “strandedness”¶
This option behaves the same as the -S option for bedtools intersect while scanning for overlaps within the “window” surrounding A. That is, overlaps in B will only be included if the B interval is on the opposite strand as the A interval.
5.1.38.8. -u Reporting the presence/absence of at least one overlapping feature¶
This option behaves the same as for bedtools intersect. That is, even if multiple overlaps exist, each A interval will only be reported once.
5.1.38.9. -c Reporting the number of overlapping features¶
This option behaves the same as for bedtools intersect. That is, it will report the count of intervals in B that overlap each A interval.
5.1.38.10. -v Reporting the absence of any overlapping features¶
This option behaves the same as for bedtools intersect. That is, it will only report those intervals in A that have have zero overlaps in B.
5.1.38.11. -header Print the header for the A file before reporting results.¶
By default, if your A file has a header, it is ignored when reporting results. This option will instead tell bedtools to first print the header for the A file prior to reporting results.

Table Of Contents
- 5.1.38. window
- 5.1.38.1. Usage and option summary
- 5.1.38.2. Default behavior
- 5.1.38.3. -w Defining a custom window size
- 5.1.38.4. -l and -r Defining assymteric windows
- 5.1.38.5. -sw Defining assymteric windows based on strand
- 5.1.38.6. -sm Enforcing matches with the same “strandedness”
- 5.1.38.7. -Sm Enforcing matches with the opposite “strandedness”
- 5.1.38.8. -u Reporting the presence/absence of at least one overlapping feature
- 5.1.38.9. -c Reporting the number of overlapping features
- 5.1.38.10. -v Reporting the absence of any overlapping features
- 5.1.38.11. -header Print the header for the A file before reporting results.