


Next: Algorithms
Up: Model
Previous: The gene model
Contents
The probability of the model has to compared to an alternative model
(in fact to all alternative models which are possible) to allow proper
Bayesian inference. This causes considerable difficulty in these
algorithms because from a algorithmical point of view we would
probably like to use an alternative model which is a single state,
like the random model in profile-HMMs, where we can simply 'log-odd'
the scored model, whereas from a biological point of view we probably
want to use a full gene predicting alternative model.
In addition we need to account for the fact that the protein HMM or
protein homolog probably does not extend over all the gene sequence,
nor in fact does the gene have to be the only gene in the DNA
sequence. This means that there are very good splice
sites/poly-pyrimidine tracts outside of the 'matched' alignment can
severely de-rail the alignment.
Basically we are in trouble with the random model parts of this
problem.
The solutions is different in the genewise21:93 compared to the genewise 6:23
algorithms. Genewise 6:23 is shown in figure 2
Figure 2:
GeneWise6:23
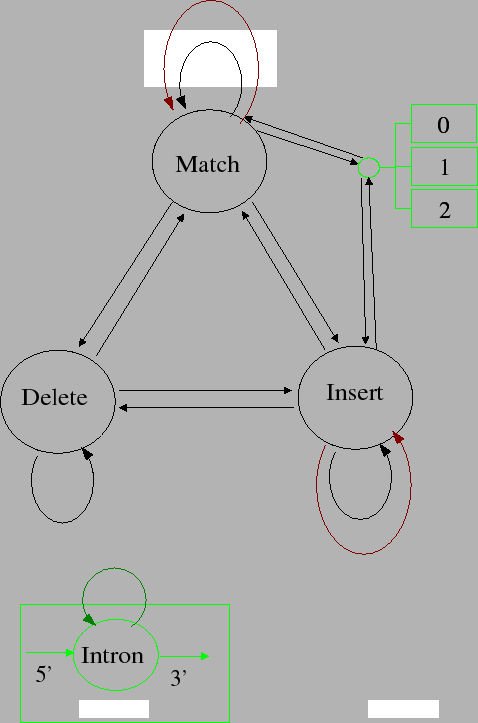 |
- In 6:23 we force the external match portions of the homology
model to be identical to the alternative model, thus cancelling each
other out. This is a pretty gross approximation and is sort of
equivalent to the intron tie'ing. It makes things algorithmically
easier... However this means a) 6:23 is nowhere near a probabilistic
model and b) you really have to used a tied intron model in 6:23
otherwise very bad edge effects (final introns being ridiculously
long) occur.
- In 21:93 we have a full probabilistic model on each side
of the homology segment. This is not reported in the -pretty output
but you can see it in the -alb output if you like. Do not trust the
gene model outside of the homology segment however. By having these
external gene model parts we can use all the gene model features safe
in the knowledge that if the homology segments do not justify the
match then the external part of the model will soak up the additional
intron/py-tract/splice site biases.
However this still does not solve the problem about what to compare it
to.
There are two approaches to the comparison
- flat
- The homology model is scored against a single state 0.25 emission
model. This is effectively 'how likely is this DNA segement has any
genes some with this homologous protein/HMM in it' for 21:93. It is,
unsurprisingly, a massive 'yes' for nearly all biological DNA, and
though a valid number in terms in bayesian inference pretty
biologically uninteresing. There is also no decent interpretation of
partial scores (ie, scores per domain).
- syn
- For synchronous model pretends that there is an alternative
model of a complete gene which is dragged into the coding part of the
gene when the homology model is in the coding part. This is not
probabilistically valid, but gives better results and interpretable
scores for partial regions, ie domain by domain. (in fact, very
similar scores to protein sequences). However I'm worried about what I
am doing It would be much better to get some mathematically
justification for this.
Next: Algorithms
Up: Model
Previous: The gene model
Contents
Eric DEVEAUD
2015-02-27