Predicting Consensus Structures from Alignment(s)
[RNA Secondary Structure Folding]
compute various properties (consensus MFE structures, partition function, Boltzmann distributed stochastic samples, ...) for RNA sequence alignments More...
Modules | |
| MFE Consensus Structures for Sequence Alignment(s) | |
| Partition Function and Base Pair Probabilities for Sequence Alignment(s) | |
| Stochastic Backtracking of Consensus Structures from Sequence Alignment(s) | |
| Local MFE consensus structures for Sequence Alignments | |
Functions | |
| int | get_mpi (char *Alseq[], int n_seq, int length, int *mini) |
| Get the mean pairwise identity in steps from ?to?(ident). | |
| float ** | readribosum (char *name) |
| Read a ribosum or other user-defined scoring matrix. | |
| float | energy_of_alistruct (const char **sequences, const char *structure, int n_seq, float *energy) |
| Calculate the free energy of a consensus structure given a set of aligned sequences. | |
| void | encode_ali_sequence (const char *sequence, short *S, short *s5, short *s3, char *ss, unsigned short *as, int circ) |
| Get arrays with encoded sequence of the alignment. | |
| void | alloc_sequence_arrays (const char **sequences, short ***S, short ***S5, short ***S3, unsigned short ***a2s, char ***Ss, int circ) |
| Allocate memory for sequence array used to deal with aligned sequences. | |
| void | free_sequence_arrays (unsigned int n_seq, short ***S, short ***S5, short ***S3, unsigned short ***a2s, char ***Ss) |
| Free the memory of the sequence arrays used to deal with aligned sequences. | |
| int | get_alipf_arrays (short ***S_p, short ***S5_p, short ***S3_p, unsigned short ***a2s_p, char ***Ss_p, FLT_OR_DBL **qb_p, FLT_OR_DBL **qm_p, FLT_OR_DBL **q1k_p, FLT_OR_DBL **qln_p, short **pscore) |
| Get pointers to (almost) all relavant arrays used in alifold's partition function computation. | |
Variables | |
| double | cv_fact |
| This variable controls the weight of the covariance term in the energy function of alignment folding algorithms. | |
| double | nc_fact |
| This variable controls the magnitude of the penalty for non-compatible sequences in the covariance term of alignment folding algorithms. | |
Detailed Description
compute various properties (consensus MFE structures, partition function, Boltzmann distributed stochastic samples, ...) for RNA sequence alignments
Consensus structures can be predicted by a modified version of the fold() algorithm that takes a set of aligned sequences instead of a single sequence. The energy function consists of the mean energy averaged over the sequences, plus a covariance term that favors pairs with consistent and compensatory mutations and penalizes pairs that cannot be formed by all structures. For details see hofacker:2002 and bernhart:2008.
Function Documentation
| int get_mpi | ( | char * | Alseq[], | |
| int | n_seq, | |||
| int | length, | |||
| int * | mini | |||
| ) |
Get the mean pairwise identity in steps from ?to?(ident).
- Parameters:
-
Alseq n_seq The number of sequences in the alignment length The length of the alignment mini
- Returns:
- The mean pairwise identity
| float energy_of_alistruct | ( | const char ** | sequences, | |
| const char * | structure, | |||
| int | n_seq, | |||
| float * | energy | |||
| ) |
Calculate the free energy of a consensus structure given a set of aligned sequences.
- Parameters:
-
sequences The NULL terminated array of sequences structure The consensus structure n_seq The number of sequences in the alignment energy A pointer to an array of at least two floats that will hold the free energies (energy[0] will contain the free energy, energy[1] will be filled with the covariance energy term)
- Returns:
- free energy in kcal/mol
| void encode_ali_sequence | ( | const char * | sequence, | |
| short * | S, | |||
| short * | s5, | |||
| short * | s3, | |||
| char * | ss, | |||
| unsigned short * | as, | |||
| int | circ | |||
| ) |
Get arrays with encoded sequence of the alignment.
this function assumes that in S, S5, s3, ss and as enough space is already allocated (size must be at least sequence length+2)
- Parameters:
-
sequence The gapped sequence from the alignment S pointer to an array that holds encoded sequence s5 pointer to an array that holds the next base 5' of alignment position i s3 pointer to an array that holds the next base 3' of alignment position i ss as circ assume the molecules to be circular instead of linear (circ=0)
| void alloc_sequence_arrays | ( | const char ** | sequences, | |
| short *** | S, | |||
| short *** | S5, | |||
| short *** | S3, | |||
| unsigned short *** | a2s, | |||
| char *** | Ss, | |||
| int | circ | |||
| ) |
Allocate memory for sequence array used to deal with aligned sequences.
Note that these arrays will also be initialized according to the sequence alignment given
- See also:
- free_sequence_arrays()
- Parameters:
-
sequences The aligned sequences S A pointer to the array of encoded sequences S5 A pointer to the array that contains the next 5' nucleotide of a sequence position S3 A pointer to the array that contains the next 3' nucleotide of a sequence position a2s A pointer to the array that contains the alignment to sequence position mapping Ss A pointer to the array that contains the ungapped sequence circ assume the molecules to be circular instead of linear (circ=0)
| void free_sequence_arrays | ( | unsigned int | n_seq, | |
| short *** | S, | |||
| short *** | S5, | |||
| short *** | S3, | |||
| unsigned short *** | a2s, | |||
| char *** | Ss | |||
| ) |
Free the memory of the sequence arrays used to deal with aligned sequences.
This function frees the memory previously allocated with alloc_sequence_arrays()
- See also:
- alloc_sequence_arrays()
- Parameters:
-
n_seq The number of aligned sequences S A pointer to the array of encoded sequences S5 A pointer to the array that contains the next 5' nucleotide of a sequence position S3 A pointer to the array that contains the next 3' nucleotide of a sequence position a2s A pointer to the array that contains the alignment to sequence position mapping Ss A pointer to the array that contains the ungapped sequence
| int get_alipf_arrays | ( | short *** | S_p, | |
| short *** | S5_p, | |||
| short *** | S3_p, | |||
| unsigned short *** | a2s_p, | |||
| char *** | Ss_p, | |||
| FLT_OR_DBL ** | qb_p, | |||
| FLT_OR_DBL ** | qm_p, | |||
| FLT_OR_DBL ** | q1k_p, | |||
| FLT_OR_DBL ** | qln_p, | |||
| short ** | pscore | |||
| ) |
Get pointers to (almost) all relavant arrays used in alifold's partition function computation.
- Note:
- To obtain meaningful pointers, call alipf_fold first!
- See also:
- pf_alifold(), alipf_circ_fold()
- Parameters:
-
S_p A pointer to the 'S' array (integer representation of nucleotides) S5_p A pointer to the 'S5' array S3_p A pointer to the 'S3' array a2s_p A pointer to the pair type matrix Ss_p A pointer to the 'Ss' array qb_p A pointer to the QB matrix qm_p A pointer to the QM matrix q1k_p A pointer to the 5' slice of the Q matrix ( 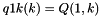 )
) qln_p A pointer to the 3' slice of the Q matrix ( 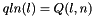 )
)
- Returns:
- Non Zero if everything went fine, 0 otherwise
Variable Documentation
| double cv_fact |
This variable controls the weight of the covariance term in the energy function of alignment folding algorithms.
Default is 1.
| double nc_fact |
This variable controls the magnitude of the penalty for non-compatible sequences in the covariance term of alignment folding algorithms.
Default is 1.
