5.1.12. flank¶
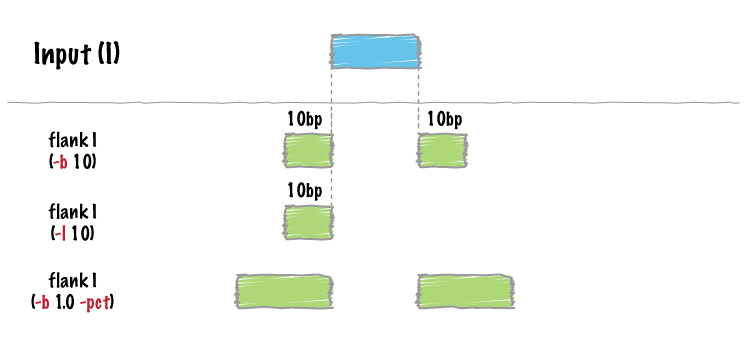
bedtools flank will create two new flanking intervals for each interval in a BED/GFF/VCF file. Note that flank will restrict the created flanking intervals to the size of the chromosome (i.e. no start < 0 and no end > chromosome size).
Note
In order to prevent creating intervals that violate chromosome boundaries, bedtools flank requires a genome file defining the length of each chromosome or contig.
See also
5.1.12.1. Usage and option summary¶
Usage:
bedtools flank [OPTIONS] -i <BED/GFF/VCF> -g <GENOME> [-b or (-l and -r)]
(or):
flankBed [OPTIONS] -i <BED/GFF/VCF> -g <GENOME> [-b or (-l and -r)]
| Option | Description |
|---|---|
| -b | Increase the BED/GFF/VCF entry by the same number base pairs in each direction. Integer. |
| -l | The number of base pairs to subtract from the start coordinate. Integer. |
| -r | The number of base pairs to add to the end coordinate. Integer. |
| -s | Define -l and -r based on strand. For example. if used, -l 500 for a negative-stranded feature, it will add 500 bp to the end coordinate. |
| -pct | Define -l and -r as a fraction of the feature’s length. E.g. if used on a 1000bp feature, -l 0.50, will add 500 bp “upstream”. Default = false. |
5.1.12.2. Default behavior¶
By default, bedtools flank will either add a fixed number of bases in each direction (-b) or an asymmetric number of bases in each direction with -l and -r.
$ cat A.bed
chr1 100 200
chr1 500 600
$ cat my.genome
chr1 1000
$ bedtools flank -i A.bed -g my.genome -b 5
chr1 95 100
chr1 200 205
chr1 495 500
chr1 600 605
$ bedtools flank -i A.bed -g my.genome -l 2 -r 3
chr1 98 100
chr1 200 203
chr1 498 500
chr1 600 603
However, if the requested number of bases exceeds the boundaries of the chromosome, bedtools flank will “clip” the feature accordingly.
$ cat A.bed
chr1 100 200
chr1 500 600
$ cat my.genome
chr1 1000
$ bedtools flank -i A.bed -g my.genome -b 800
chr1 0 100
chr1 200 1000
chr1 0 500
chr1 600 1000
5.1.12.3. -pct Resizing features by a given fraction¶
bedtools flank will optionally create flanking intervals whose size is user-specified fraction of the original interval.
For example:
$ cat A.bed
chr1 100 200
chr1 500 700
################################################################
# note the flanking intervals from the second record in A.bed
# are 20bp whereas the flanking intervals from the first record
# are only 10bp
################################################################
$ bedtools flank -i A.bed -g my.genome -b 0.1 -pct
chr1 90 100
chr1 200 210
chr1 480 500
chr1 700 720
