5.1.23. merge¶
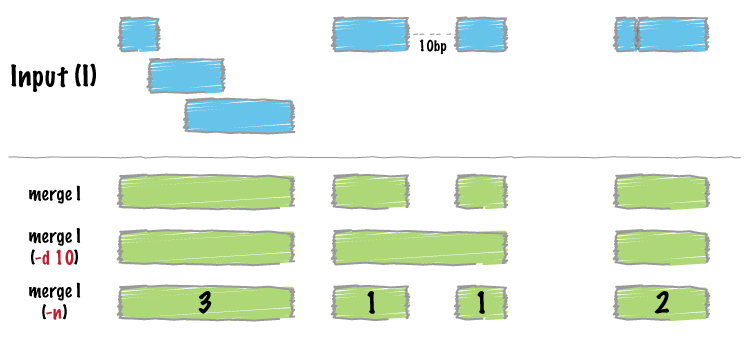
bedtools merge combines overlapping or “book-ended” features in an interval file into a single feature which spans all of the combined features.
Note
bedtools merge requires that you presort your data by chromosome and then by start position (e.g., sort -k1,1 -k2,2n in.bed > in.sorted.bed for BED files).
See also
5.1.23.1. Usage and option summary¶
Usage:
bedtools merge [OPTIONS] -i <BED/GFF/VCF>
(or):
mergeBed [OPTIONS] -i <BED/GFF/VCF>
| Option | Description |
|---|---|
| -s | Force strandedness. That is, only merge features that are the same strand. By default, this is disabled. |
| -n | Report the number of BED entries that were merged. 1 is reported if no merging occurred. |
| -d | Maximum distance between features allowed for features to be merged. Default is 0. That is, overlapping and/or book-ended features are merged. |
| -nms | Report the names of the merged features separated by commas. Change delimiter with -delim |
| -scores | Report the scores of the merged features.
Specify one of the following options for reporting scores:
sum, min, max,
mean, median, mode, antimode,
collapse (i.e., print a semicolon-separated list)
|
| -delim | Specify a custom delimiter for the -nms and -scores concat options
Example: -delim "|"
Default: ";"
|
5.1.23.2. Default behavior¶
By default, bedtools merge combines overlapping (by at least 1 bp) and/or bookended intervals into a single, “flattened” or “merged” interval.
$ cat A.bed
chr1 100 200
chr1 180 250
chr1 250 500
chr1 501 1000
$ bedtools merge -i A.bed
chr1 100 500
chr1 501 1000
5.1.23.3. -s Enforcing “strandedness”¶
The -s option will only merge intervals that are overlapping/bookended and are on the same strand.
$ cat A.bed
chr1 100 200 a1 1 +
chr1 180 250 a2 2 +
chr1 250 500 a3 3 -
chr1 501 1000 a4 4 +
$ bedtools merge -i A.bed -s
chr1 100 250 +
chr1 501 1000 +
chr1 250 500 -
5.1.23.4. -n Reporting the number of features that were merged¶
The -n option will report the number of features that were combined from the original file in order to make the newly merged feature. If a feature in the original file was not merged with any other features, a “1” is reported.
$ cat A.bed
chr1 100 200
chr1 180 250
chr1 250 500
chr1 501 1000
$ bedtools merge -i A.bed -n
chr1 100 500 3
chr1 501 1000 1
5.1.23.5. -d Controlling how close two features must be in order to merge¶
By default, only overlapping or book-ended features are combined into a new feature. However, one can force merge to combine more distant features with the -d option. For example, were one to set -d to 1000, any features that overlap or are within 1000 base pairs of one another will be combined.
$ cat A.bed
chr1 100 200
chr1 501 1000
$ bedtools merge -i A.bed
chr1 100 200
chr1 501 1000
$ bedtools merge -i A.bed -d 1000
chr1 100 200 1000
5.1.23.6. -nms Reporting the names of the features that were merged¶
Occasionally, one might like to know that names of the features that were merged into a new feature. The -nms option will add an extra column to the merge output which lists (separated by semicolons) the names of the merged features.
$ cat A.bed
chr1 100 200 A1
chr1 150 300 A2
chr1 250 500 A3
$ bedtools merge -i A.bed -nms
chr1 100 500 A1,A2,A3
5.1.23.7. -scores Reporting the scores of the features that were merged¶
Similarly, we might like to know that scores of the features that were merged into a new feature. Enter the -scores option. One can specify how the scores from each overlapping interval should be reported.
$ cat A.bed
chr1 100 200 A1 1
chr1 150 300 A2 2
chr1 250 500 A3 3
$ bedtools merge -i A.bed -scores mean
chr1 100 500 2
$ bedtools merge -i A.bed -scores max
chr1 100 500 3
$ bedtools merge -i A.bed -scores collapse
chr1 100 500 1,2,3
5.1.23.8. -delim Change the delimiter for -nms and -scores collapse¶
One can override the use of a comma as the delimiter for the -nms and -scores collapse options via the use of the -delim option.
$ cat A.bed
chr1 100 200 A1
chr1 150 300 A2
chr1 250 500 A3
Compare:
$ bedtools merge -i A.bed -nms
chr1 100 500 A1,A2,A3
to:
$ bedtools merge -i A.bed -nms -delim "|"
chr1 100 500 A1|A2|A3

Table Of Contents
- 5.1.23. merge
- 5.1.23.1. Usage and option summary
- 5.1.23.2. Default behavior
- 5.1.23.3. -s Enforcing “strandedness”
- 5.1.23.4. -n Reporting the number of features that were merged
- 5.1.23.5. -d Controlling how close two features must be in order to merge
- 5.1.23.6. -nms Reporting the names of the features that were merged
- 5.1.23.7. -scores Reporting the scores of the features that were merged
- 5.1.23.8. -delim Change the delimiter for -nms and -scores collapse