Folding Routines - Functions for Folding RNA Secondary Structures
Table of Contents
- Calculating Minimum Free Energy Structures
- Calculating Partition Functions and Pair Probabilities
- Searching for Predefined Structures
- Enumerating Suboptimal Structures
- Predicting hybridization structures of two molecules
- Predicting local structures of large sequences
- Predicting Consensus Structures from Alignment
- Global Variables for the Folding Routines
- Reading Energy Parameters from File
Calculating Minimum Free Energy Structures
The library provides a fast dynamic programming minimum free energy folding algorithm as described by Zuker & Stiegler (1981).
Associated functions are:
float fold (char* sequence, char* structure);
Compute minimum free energy and an appropriate secondary structure of an RNA sequence.
float circfold (char* sequence, char* structure);
Compute minimum free energy and an appropriate secondary structure of a circular RNA sequence.
float energy_of_structure(const char *string,
const char *structure,
int verbosity_level);
Calculate the free energy of an already folded RNA using global model detail settings.
float energy_of_circ_structure( const char *string,
const char *structure,
int verbosity_level);
Calculate the free energy of an already folded circular RNA.
void update_fold_params(void);
Recalculate energy parameters.
void free_arrays(void);
Free arrays for mfe folding.
- See also:
- fold.h, cofold.h, 2Dfold.h, Lfold.h, alifold.h and subopt.h for a complete list of available functions.
Table of Contents
Calculating Partition Functions and Pair Probabilities
Instead of the minimum free energy structure the partition function of all possible structures and from that the pairing probability for every possible pair can be calculated, using a dynamic programming algorithm as described by McCaskill (1990). The following functions are provided:
float pf_fold ( char* sequence,
char* structure)
Compute the partition function 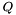 of an RNA sequence.
of an RNA sequence.
void free_pf_arrays (void)
Free arrays for the partition function recursions.
void update_pf_params (int length)
Recalculate energy parameters.
char *get_centroid_struct_pl( int length,
double *dist,
plist *pl);
Get the centroid structure of the ensemble.
char *get_centroid_struct_pr( int length,
double *dist,
double *pr);
Get the centroid structure of the ensemble.
double mean_bp_distance_pr( int length,
double *pr);
Get the mean base pair distance in the thermodynamic ensemble.
- See also:
- part_func.h, part_func_co.h, part_func_up.h, 2Dpfold.h, LPfold.h, alifold.h and MEA.h for a complete list of available functions.
Table of Contents
Searching for Predefined Structures
We provide two functions that search for sequences with a given structure, thereby inverting the folding routines.
float inverse_fold (char *start,
char *target)
Find sequences with predefined structure.
float inverse_pf_fold ( char *start,
char *target)
Find sequence that maximizes probability of a predefined structure.
The following global variables define the behavior or show the results of the inverse folding routines:
char *symbolset
This global variable points to the allowed bases, initially "AUGC".
- See also:
- inverse.h for more details and a complete list of available functions.
Table of Contents
Enumerating Suboptimal Structures
SOLUTION *subopt (char *sequence,
char *constraint,
int *delta,
FILE *fp)
Returns list of subopt structures or writes to fp.
SOLUTION *subopt_circ ( char *sequence,
char *constraint,
int *delta,
FILE *fp)
Returns list of circular subopt structures or writes to fp.
SOLUTION *zukersubopt(const char *string);
Compute Zuker type suboptimal structures.
char *TwoDpfold_pbacktrack ( TwoDpfold_vars *vars,
unsigned int d1,
unsigned int d2)
Sample secondary structure representatives from a set of distance classes according to their Boltzmann probability.
char *alipbacktrack (double *prob)
Sample a consensus secondary structure from the Boltzmann ensemble according its probability
.
char *pbacktrack(char *sequence);
Sample a secondary structure from the Boltzmann ensemble according its probability
.
char *pbacktrack_circ(char *sequence);
Sample a secondary structure of a circular RNA from the Boltzmann ensemble according its probability.
- See also:
- subopt.h, part_func.h, alifold.h and 2Dpfold.h for more detailed descriptions
Table of Contents
Predicting hybridization structures of two molecules
The function of an RNA molecule often depends on its interaction with other RNAs. The following routines therefore allow to predict structures formed by two RNA molecules upon hybridization.
One approach to co-folding two RNAs consists of concatenating the two sequences and keeping track of the concatenation point in all energy evaluations. Correspondingly, many of the cofold() and co_pf_fold() routines below take one sequence string as argument and use the the global variable cut_point to mark the concatenation point. Note that while the RNAcofold program uses the '&' character to mark the chain break in its input, you should not use an '&' when using the library routines (set cut_point instead).
In a second approach to co-folding two RNAs, cofolding is seen as a stepwise process. In the first step the probability of an unpaired region is calculated and in a second step this probability of an unpaired region is multiplied with the probability of an interaction between the two RNAs. This approach is implemented for the interaction between a long target sequence and a short ligand RNA. Function pf_unstru() calculates the partition function over all unpaired regions in the input sequence. Function pf_interact(), which calculates the partition function over all possible interactions between two sequences, needs both sequence as separate strings as input.
int cut_point
Marks the position (starting from 1) of the first nucleotide of the second molecule within the concatenated sequence.
float cofold (char *sequence,
char *structure)
Compute the minimum free energy of two interacting RNA molecules.
void free_co_arrays (void)
Free memory occupied by cofold().
Partition Function Cofolding
To simplify the implementation the partition function computation is done internally in a null model that does not include the duplex initiation energy, i.e. the entropic penalty for producing a dimer from two monomers). The resulting free energies and pair probabilities are initially relative to that null model. In a second step the free energies can be corrected to include the dimerization penalty, and the pair probabilities can be divided into the conditional pair probabilities given that a re dimer is formed or not formed.
cofoldF co_pf_fold( char *sequence,
char *structure);
Calculate partition function and base pair probabilities.
void free_co_pf_arrays(void);
Free the memory occupied by co_pf_fold().
Cofolding all Dimeres, Concentrations
After computing the partition functions of all possible dimeres one can compute the probabilities of base pairs, the concentrations out of start concentrations and sofar and soaway.
void compute_probabilities(
double FAB,
double FEA,
double FEB,
struct plist *prAB,
struct plist *prA,
struct plist *prB,
int Alength)
Compute Boltzmann probabilities of dimerization without homodimers.
ConcEnt *get_concentrations(double FEAB,
double FEAA,
double FEBB,
double FEA,
double FEB,
double * startconc)
Given two start monomer concentrations a and b, compute the concentrations in thermodynamic equilibrium of all dimers and the monomers.
Partition Function Cofolding as a stepwise process
In this approach to cofolding the interaction between two RNA molecules is seen as a stepwise process. In a first step, the target molecule has to adopt a structure in which a binding site is accessible. In a second step, the ligand molecule will hybridize with a region accessible to an interaction. Consequently the algorithm is designed as a two step process: The first step is the calculation of the probability that a region within the target is unpaired, or equivalently, the calculation of the free energy needed to expose a region. In the second step we compute the free energy of an interaction for every possible binding site. Associated functions are:
pu_contrib *pf_unstru ( char *sequence,
int max_w)
Calculate the partition function over all unpaired regions of a maximal length.
void free_pu_contrib_struct (pu_contrib *pu)
Frees the output of function pf_unstru().
interact *pf_interact(
const char *s1,
const char *s2,
pu_contrib *p_c,
pu_contrib *p_c2,
int max_w,
char *cstruc,
int incr3,
int incr5)
Calculates the probability of a local interaction between two sequences.
void free_interact (interact *pin)
Frees the output of function pf_interact().
- See also:
- cofold.h, part_func_co.h and part_func_up.h for more details
Table of Contents
Predicting local structures of large sequences
Local structures can be predicted by a modified version of the fold() algorithm that restricts the span of all base pairs.
float Lfold ( const char *string,
char *structure,
int maxdist)
The local analog to fold().
float aliLfold( const char **strings,
char *structure,
int maxdist)
float Lfoldz (const char *string,
char *structure,
int maxdist,
int zsc,
double min_z)
plist *pfl_fold (
char *sequence,
int winSize,
int pairSize,
float cutoffb,
double **pU,
struct plist **dpp2,
FILE *pUfp,
FILE *spup)
Compute partition functions for locally stable secondary structures (berni! update me).
Table of Contents
Predicting Consensus Structures from Alignment
Consensus structures can be predicted by a modified version of the fold() algorithm that takes a set of aligned sequences instead of a single sequence. The energy function consists of the mean energy averaged over the sequences, plus a covariance term that favors pairs with consistent and compensatory mutations and penalizes pairs that cannot be formed by all structures. For details see Hofacker (2002).
float alifold (const char **strings,
char *structure)
Compute MFE and according consensus structure of an alignment of sequences.
float circalifold (const char **strings,
char *structure)
Compute MFE and according structure of an alignment of sequences assuming the sequences are circular instead of linear.
void free_alifold_arrays (void)
Free the memory occupied by MFE alifold functions.
float energy_of_alistruct (
const char **sequences,
const char *structure,
int n_seq,
float *energy)
Calculate the free energy of a consensus structure given a set of aligned sequences.
struct pair_info
A base pair info structure.
double cv_fact
This variable controls the weight of the covariance term in the energy function of alignment folding algorithms.
double nc_fact
This variable controls the magnitude of the penalty for non-compatible sequences in the covariance term of alignment folding algorithms.
- See also:
- alifold.h for more details
Table of Contents
Global Variables for the Folding Routines
The following global variables change the behavior the folding algorithms or contain additional information after folding.
int noGU
Global switch to forbid/allow GU base pairs at all.
int no_closingGU
GU allowed only inside stacks if set to 1.
int noLonelyPairs
Global switch to avoid/allow helices of length 1.
int tetra_loop
Include special stabilizing energies for some tri-, tetra- and hexa-loops;.
int energy_set
0 = BP; 1=any mit GC; 2=any mit AU-parameter
float temperature
Rescale energy parameters to a temperature in degC.
int dangles
Switch the energy model for dangling end contributions (0, 1, 2, 3).
char *nonstandards
contains allowed non standard base pairs
int cut_point
Marks the position (starting from 1) of the first nucleotide of the second molecule within the concatenated sequence.
float pf_scale
A scaling factor used by pf_fold() to avoid overflows.
int fold_constrained
Global switch to activate/deactivate folding with structure constraints.
int do_backtrack
do backtracking, i.e.
char backtrack_type
A backtrack array marker for inverse_fold().
include fold_vars.h if you want to change any of these variables from their defaults.
- See also:
- fold_vars.h for a more complete and detailed description of all global variables and how to use them
Table of Contents
Reading Energy Parameters from File
A default set of parameters, identical to the one described in Mathews et.al. (2004), is compiled into the library.
Alternately, parameters can be read from and written to a file.
void read_parameter_file (const char fname[])
Read energy parameters from a file.
void write_parameter_file (const char fname[])
Write energy parameters to a file.
To preserve some backward compatibility the RNAlib also provides functions to convert energy parameter files from the format used in version 1.4-1.8 into the new format used since version 2.0
void convert_parameter_file (
const char *iname,
const char *oname,
unsigned int options)
Convert/dump a Vienna 1.8.4 formatted energy parameter file.
- See also:
- read_epars.h and convert_epars.h for detailed description of the available functions
Table of Contents
